Long QT Syndrome (LQTS)
Overview
Long QT syndrome is the most prevalent cardiac channelopathy, characterised by prolongation of the QT interval on ECG due to abnormal cardiac repolarization. It predisposes to torsades de pointes (TdP), ventricular fibrillation, and sudden cardiac death in the absence of structural heart disease. LQTS encompasses 17 genetic subtypes; the three major forms — LQT1 (KCNQ1), LQT2 (KCNH2), and LQT3 (SCN5A) — account for ~90% of genotype-positive cases. Clinical presentation ranges from asymptomatic QT prolongation to syncope, aborted cardiac arrest, and SCD, often triggered by exercise, emotion, or auditory stimuli depending on subtype. Management is stepwise: lifestyle modification + beta-blockers as the foundation, escalating to mexiletine (LQT3), LCSD, and ICD for higher-risk patients.
Key Facts
Epidemiology
- Prevalence: Documented 1:2000 live births (prospective study, n=44,000 infants); actual prevalence probably higher because genotype-positive–phenotype-negative persons were excluded. (sources/lqts-nejm-2025, rating: very high; sources/channelopathies-jaha-2025, rating: high)
- Arrhythmic risk by age: Highest in childhood; decreases with age; patients >60 years have attenuated risk. (sources/channelopathies-jaha-2025)
- Sex-based risk: Males aged 10–12 have 4× higher arrhythmic risk than females. From ages 18–40, risk reverses: women 11% vs. men 2%. Sex hormones modulate IKr — oestrogen inhibits (↑QT), testosterone potentiates (↓QT). Postpartum period carries elevated risk. (sources/channelopathies-jaha-2025)
- Asymptomatic carrier risk: Family members with a confirmed pathogenic LQTS variant but normal QTc carry a tenfold increased risk of cardiac events compared to non-carriers — justifying variant analysis in all first-degree relatives regardless of QTc. (sources/arrhythmia-genetics-mgenetik-2025, rating: high)
- Drug-induced QTc prolongation: Only 10–15% of cases represent unmasked hidden LQTS. (sources/arrhythmia-genetics-mgenetik-2025) — See Contradictions: the AHA 2020 statement cites ~30% carrying latent LQTS variants in drug-induced TdP specifically. (sources/drug-arrhythmia-aha-2020, rating: very high)
Genetics
Major Subtypes
- Three major subtypes account for ~90% of genotype-positive cases: LQT1 (KCNQ1, ~50%), LQT2 (KCNH2, ~40%), LQT3 (SCN5A, ~10%), all identified 1995–1996. (sources/lqts-nejm-2025, rating: very high) — Older symptomatic-cohort registries report lower rates (LQT1 30–35%, LQT2 25–30%), reflecting ascertainment bias. (sources/channelopathies-jaha-2025, rating: high)
- Overall variant detection rate: 70–80% across all LQTS subtypes. (sources/arrhythmia-genetics-mgenetik-2025)
- Notable syndromal subtypes: Timothy syndrome (LQT8, CACNA1C GOF) — rare, highly malignant, autism + webbed digits + immune deficiency; Jervell and Lange-Nielsen syndrome (KCNQ1/KCNE1 homozygous/compound het) — autosomal recessive, sensorineural deafness, severe LQTS, high mortality; Andersen-Tawil syndrome (LQT7, KCNJ2 LOF) — periodic paralysis + arrhythmias + dysmorphic features, see entities/Andersen-Tawil-Syndrome. (sources/channelopathies-jaha-2025)
- High-risk genetic features: Transmembrane pore variants in KCNH2 carry the greatest relative cardiac event risk within LQT2; dominant-negative variants carry greater risk than haploinsufficiency variants. (sources/channelopathies-jaha-2025, sources/lqts-jaccep-2022)
ClinGen Gene-Disease Validity (2026)
- Definitive LQTS genes: KCNQ1 (LQT1), KCNH2 (LQT2), CALM1/2/3 (calmodulinopathy), CACNA1C (Timothy syndrome/LQT8). TRDN has strong evidence for autosomal recessive LQTS/CPVT5 — important in recessive unexplained neonatal arrhythmia. (sources/clingen-summary-2026-05-09, rating: high)
- SCN5A — broad reclassification: ClinGen now uses "SCN5A-related cardiac rhythm disorder" (definitive 10/08/2025), encompassing LQT3, Brugada syndrome, DCM, and conduction disease under one entity rather than a separate LQTS designation. (sources/clingen-summary-2026-05-09)
- Disputed or limited evidence: SCN4B (LQT10) — disputed; variants must not be classified P/LP for LQTS. KCNE2 — disputed and removed (Adler 2020). CAV3 (LQT9) and KCNJ2 (LQT7) — limited evidence for isolated LQTS; primarily relevant for Andersen-Tawil syndrome. (sources/clingen-summary-2026-05-09, sources/arrhythmia-genetics-mgenetik-2025)
- See concepts/ClinGen-Gene-Disease-Validity for full framework.
Calmodulinopathy LQTS
- Clinical phenotype: Near-complete penetrance despite heterozygosity (mutant CaMs compete equally for the pre-bound Cav1.2 pool). Extreme QTc (often >600 ms), neonatal or early-childhood onset, high SCD risk; predominantly de novo — negative family history does not exclude. Neonatal LQTS with QTc >600 ms and no SCN5A/KCNQ1/KCNH2 cause → CALM1/2/3 sequencing mandatory. (sources/arrhythmia-genetics-mgenetik-2025, sources/clingen-summary-2026-05-09, sources/CALM-FCVM-2018, rating: high)
- Mechanism: EF-hand III/IV mutations reduce C-lobe Ca²⁺ affinity → failed CDI of Cav1.2 → ICaL GOF → APD prolongation → EADs → TdP/VF. (sources/CALM-FCVM-2018)
- Therapy: Verapamil (ICaL blockade) reverses QT prolongation in hiPSC-CM models; selective sustained ICaL blockade is the mechanistic therapeutic target. (sources/CALM-FCVM-2018)
- See concepts/Calmodulinopathy for full mechanism and mutation catalogue.
Modifier Genes and Polygenic Risk
- NOS1AP: The most validated LQTS modifier gene (GWAS-derived). Common noncoding variants associated with elevated life-threatening arrhythmia risk in LQT1 (South Africa) and LQT2 (Netherlands) cohorts; also modulates drug-induced LQTS and post-AMI SCD risk. See entities/NOS1AP. (sources/modifier-genes-scd-ehj-2018, rating: high)
- Actionable coding modifiers: KCNE1-D85N — predisposes to congenital and drug-induced LQTS; sex-specific (prolongs QTc in male LQT1 carriers); avoid IKr-blocking drugs even if the sole finding. KCNH2-K897T — can convert latent LQT2 to symptomatic; also aggravates LQT1 at maximal exercise. (sources/modifier-genes-scd-ehj-2018)
- Polygenic risk: Common SNPs explain ~15% of LQTS susceptibility variance; PRS top decile adds ~8.7 ms to QTc (comparable to some monogenic variants). diLQT PRS explains ~30% of QTc variance during drug exposure (OR 1.34/SD for drug-induced LQTS). (sources/repolarisation-jaccep-2023, sources/gwas-arrhythmias-cmp-genes-2025, rating: high)
- 75% of marked unexplained QTc prolongation is non-genetic: Most patients lack both a high PRS and an identifiable rare variant — drugs, electrolytes, and autonomic tone dominate. (sources/gwas-arrhythmias-cmp-genes-2025)
- See concepts/Modifier-Genes, concepts/Polygenic-Risk-Score, and concepts/GWAS-Cardiac-Genetics for methodology.
Pathophysiology
Ion Channel Mechanisms by Subtype
- LQT1 and LQT2: Loss-of-function in KCNQ1 (IKs) and KCNH2 (IKr) respectively reduces repolarizing K⁺ currents → delayed repolarization → prolonged QT. (sources/channelopathies-jaha-2025, rating: high)
- LQT3: SCN5A gain-of-function → persistent (late) Na⁺ influx (INaLate) and/or increased window current during plateau → prolonged QT. Both residual currents amplify at slow heart rates (longer plateau) — hallmark: bradycardia-dependent QT prolongation, normalising at faster rates. Arrhythmic events predominantly at rest/sleep; first events in LQT3 more likely lethal; onset typically after puberty. (sources/channelopathies-jaha-2025, sources/scn5a-jaccep-2018)
- LQT4 (ANKB): ANK2 (ankyrin-B) loss-of-function disrupts subcellular targeting of NCX1, Na⁺/K⁺-ATPase, and IP3 receptor → mislocalisation of NCX1 away from t-tubular Ca²⁺ release sites → altered Ca²⁺ handling → APD prolongation. One of few non-channel proteins causing LQTS. (sources/membrane-potential-physrev-2021, rating: very high)
- LQT5 (KCNE1) and LQT6 (KCNE2): KCNE1 (minK) and KCNE2 (MiRP1) are β-subunits co-assembling with KCNQ1 (IKs) and KCNH2 (IKr) respectively. LOF in either reduces the associated current; KCNE2 (LQT6) disputed by ClinGen 2020. (sources/membrane-potential-physrev-2021)
- LQT8 (Timothy syndrome, CACNA1C): GOF in Cav1.2 → increased inward ICa,L → prolonged plateau → QT prolongation; impaired inactivation → Ca²⁺ overload → severe multisystem phenotype (autism, bradycardia, webbed digits). (sources/channelopathies-jaha-2025)
- LQT9 (CAV3) and LQT12 (SNTA1): Mutations in caveolin-3 or α1-syntrophin → indirect INaLate augmentation via disrupted Nav1.5 macromolecular complex → same functional consequence as LQT3 GOF. (sources/membrane-potential-physrev-2021)
- Genotype-specific T-wave morphology: IKs (KCNQ1) predominates in subepicardium and RV; nearly absent at rest → LQT1 shows minimal baseline QT prolongation that unmasks with exercise. IKr (KCNH2) prominent at low heart rates → LQT2 prolonged at baseline; IKr reduction causes greater LV APD prolongation → characteristic bifid T-wave. SCN5A GOF prolongs phase 2 → delayed T-wave onset with long ST-segment in LQT3 regardless of heart rate. (sources/repolarisation-jaccep-2023, rating: high)
Final Common Pathway (TdP Generation)
- Both K⁺ LOF and Na⁺/Ca²⁺ GOF converge on prolonged APD → M-cell APD prolongation disproportionate to subepicardium → transmural dispersion of repolarization (TDR) → EAD in mid-myocardium → triggered beat on T-wave → re-entry into dispersed substrate → TdP. (sources/channelopathies-jaha-2025, sources/membrane-potential-physrev-2021)
- Repolarization reserve: Normal repolarization depends on multiple redundant ion currents (IKr, IKs, IK1, INa-L). When one mechanism is already perturbed by a latent LQTS variant, the addition of an IKr-blocking drug depletes repolarization reserve and unmasks TdP risk — explaining why ~10–20% of drug-induced TdP patients carry LQTS gene variants. (sources/repolarisation-jaccep-2023)
Sex Hormones and Circadian Variation
- Oestradiol inhibits IKr and potentiates ICa,L → net APD prolongation → female QT ~10–12 ms longer than male baseline, with higher arrhythmic risk at ages 18–40. Testosterone upregulates IKr and IKs → shorter APD in males; pubescent males experience a QTc shortening absent in females, explaining the cross-over in arrhythmic risk around puberty. Progesterone upregulates IKs and has antiarrhythmic properties — its postpartum fall explains heightened LQT2 arrhythmic risk in the 9-month postpartum period. (sources/membrane-potential-physrev-2021, sources/lqts-pregnancy-medicina-2022, rating: medium)
- Circadian APD amplification (LQT2): Kcnh2 is circadian-regulated (BMAL1/CLOCK). Wild-type Kv11.1 protein t½ ~12 h blunts protein oscillation amplitude. KCNH2 mutations can shorten Kv11.1 t½ to <6 h → two simultaneous consequences: (1) lower steady-state Kv11.1 protein → less IKr → longer baseline APD; and (2) larger circadian APD swing → potential clustering of arrhythmic events at specific times of day. (sources/circadian-scd-jmcc-2025, rating: high)
Diagnosis
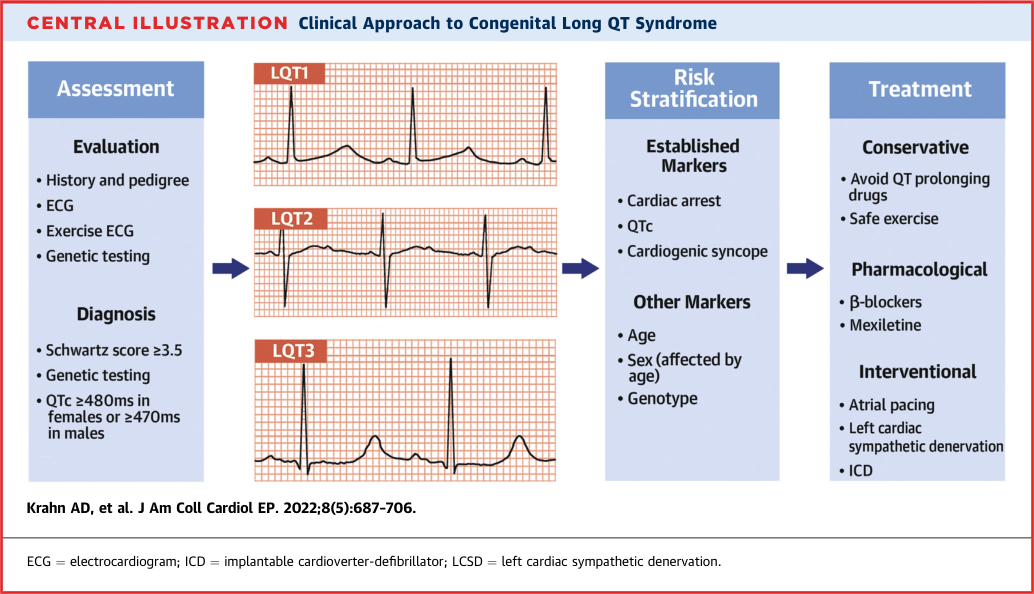
- ESC 2022 Class I diagnostic criteria (any one of):
- QTc ≥480 ms on repeated 12-lead ECGs
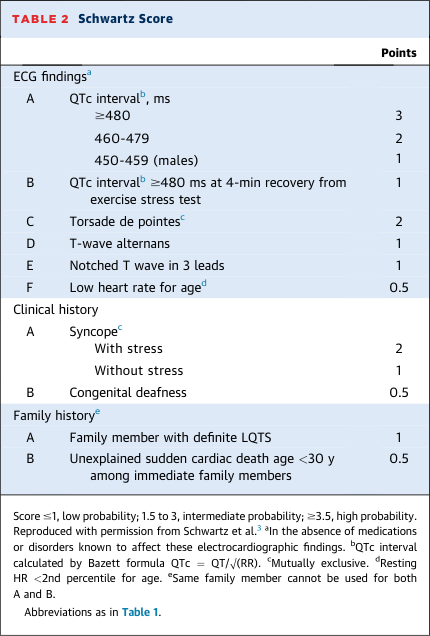
- Schwartz Score ≥3.5 points
- Pathogenic variant in an LQTS gene, irrespective of QTc duration
- (sources/VA-SCD-ESC-2022, rating: very high)
- HRS/EHRA/APHRS consensus diagnostic criteria (any one of): Schwartz score ≥3.5; pathogenic variant; repeated QTc ≥500 ms in absence of QT-prolonging drugs. Schwartz score ≥3.5 has 99% specificity, 19–36% sensitivity. (sources/lqts-jaccep-2022, rating: high)
- Apparent acquired LQTS may be congenital: ~25% of patients with apparent drug-induced LQTS harboured a pathogenic LQTS gene variant in an international cohort. All patients with drug-induced QT prolongation should be re-evaluated after drug cessation. (sources/lqts-jaccep-2022)
- Clinical QTc context: In 1,710 LQTS cases, mean QTc was 471±45 ms; 47% of LQT1, 36% of LQT2, and 35% of LQT3 patients had QTc <460 ms — confirming QTc alone is insufficient for exclusion. (sources/arrhythmia-genetics-mgenetik-2025)
- Diagnostic workup: 12-lead ECG, Schwartz Score, exercise treadmill test (QTc paradoxically shortens in LQT1 on exercise; fails to shorten in LQT2), 24-hr ambulatory ECG; genetic testing for high clinical suspicion with negative workup. (sources/channelopathies-jaha-2025)
- Genetic testing: Class I per EHRA/HRS/APHRS/LAHRS consensus; a pathogenic genetic result is part of the diagnostic score. (sources/arrhythmia-genetics-mgenetik-2025)
- AHA 2020 specific testing thresholds: Genetic testing recommended for strong clinical index of suspicion for LQTS; also for idiopathic QT prolongation with QTc >480 ms (prepuberty) or >500 ms (adults), even when asymptomatic. (sources/genetic-test-aha-2020, rating: high)
- Epinephrine challenge: NOT recommended (Class III) for routine diagnosis. EPS: NOT recommended (Class III). (sources/VA-SCD-ESC-2022)
- AI-ECG: Up to 40% of LQTS patients have QTc <450 ms at baseline. Deep learning models have achieved AUC 0.741 for identifying concealed LQTS from the 12-lead ECG and can discriminate LQT1 from LQT2. Input occlusion reveals mechanistic T-wave footprints corresponding to predicted IKr inhibition effects. (sources/repolarisation-jaccep-2023)
ECG Patterns by Subtype (LQT1–3)
The three major subtypes produce distinct T-wave signatures reflecting the regional distribution of IKs, IKr, and INaLate across the ventricular wall. These patterns support genotype prediction before genetic results are available. (sources/repolarisation-jaccep-2023, sources/lqts-jaccep-2022, rating: high)
LQT1 (KCNQ1 — IKs loss-of-function)
- T-wave: Broad-based, symmetrically prolonged, smooth upslope; normal to mildly increased amplitude
- Baseline QT: Minimal prolongation at rest — IKs is nearly absent at resting heart rates, so KCNQ1 mutations cause little baseline change; QT prolongation unmasks dramatically with sympathetic activation when IKs becomes the dominant repolarising current
- Dynamic behaviour: QTc increases during exercise, shortens gradually during recovery (opposite of normal physiology) — exercise paradoxically prolongs QT and is the primary arrhythmic trigger
- Trigger: Exercise, swimming (catecholamine surge)
LQT2 (KCNH2 — IKr loss-of-function)
- T-wave: Low-amplitude, bifid or notched (two distinct peaks, or a prominent notch on the descent); most characteristic pattern across all three subtypes
- Baseline QT: Prolonged at rest — IKr is the dominant repolarising current at slow heart rates; mutations cause resting QT prolongation without adrenergic challenge
- Mechanism of bifid T-wave: IKr reduction prolongs APD more in the LV than in the RV (where IKs partially compensates) → interventricular and transmural repolarisation disparity → two-phase T-wave morphology (sources/repolarisation-jaccep-2023)
- Dynamic behaviour: QTc shortens during exercise, then gradually re-lengthens during recovery — characteristically opposite to LQT1
- Trigger: Sudden auditory stimuli (startling), emotion, postpartum period
LQT3 (SCN5A — INaLate gain-of-function)
- T-wave: Late-onset peaked or asymmetric biphasic T-wave, preceded by a long isoelectric or flat ST-segment; abrupt upslope with relatively narrow peak
- ECG hallmark: Prolonged ST-segment (extended plateau) → delayed T-wave onset — the isoelectric gap between QRS and T-wave is visually distinct from LQT1/LQT2
- Dynamic behaviour: QTc worsens at slow heart rates (longer diastolic interval allows greater INaLate accumulation); does NOT shorten appropriately with faster rates; ambulatory ECGs often reveal more pronounced QTc prolongation during sleep than on a resting daytime ECG
- Trigger: Rest, sleep (bradycardia-dependent)
Shared features and caveats
- Giant T-U waves in any subtype herald impending TdP — a widened, prominent terminal deflection (T-U fusion) immediately precedes short-coupled VPBs that trigger the arrhythmia. (sources/lqts-jaccep-2022, rating: high)
- Pattern overlap is common; T-wave morphology is insufficient alone for genotype assignment. 36% of LQT2 and 35% of LQT3 patients have QTc <460 ms on a resting ECG — normal T-wave morphology does not exclude LQTS. (sources/arrhythmia-genetics-mgenetik-2025, rating: high)
- A single resting ECG captures one heart rate snapshot; serial ECGs at different rates (including 4-minute post-exercise recovery) improve both sensitivity and genotype differentiation. (sources/lqts-jaccep-2022)
Provocation Testing Thresholds
- Exercise testing (QTc at 4-minute recovery): QTc ≥445 ms = 90% sensitivity and 90% specificity for LQTS; QTc ≥480 ms = 36% sensitivity and 100% specificity (incorporated into updated Schwartz score). Validated for LQT1 and LQT2 in adults. (sources/lqts-jaccep-2022, rating: high)
- Genotype differentiation by exercise: LQT1 → QTc increases during exercise, gradually shortens during recovery. LQT2 → QTc shortens during exercise, gradually lengthens during recovery. Differences at peak exercise or 1-min recovery can differentiate subtypes. (sources/lqts-jaccep-2022)
- Stand-up (orthostatic) test: QTc stretch ≥490 ms (maximal QT stretch = T-wave end approaches next P-wave during RR shortening) → 92% sensitivity, 79% specificity. Not validated in paediatric patients — healthy children may increase QTc by up to 80 ms on standing. (sources/lqts-jaccep-2022)
- Paediatric exercise recovery: Optimal threshold QTc >460 ms at 7-minute recovery (rather than 4-min) in children with LQTS. (sources/lqts-jaccep-2022)
Risk Stratification
- 1-2-3 LQTS Risk Calculator: Estimates arrhythmic risk before therapy initiation; recommended prior to ICD decisions: Class IIa (ESC 2022). (sources/VA-SCD-ESC-2022, rating: very high)
- ICD in asymptomatic high-risk LQTS (per 1-2-3 LQTS Risk calculator) in addition to genotype-specific therapies: Class IIb. (sources/VA-SCD-ESC-2022)
- Trigger patterns inform risk stratification: LQT1 events triggered by exercise/swimming; LQT2 by auditory stimuli/emotion/postpartum; LQT3 at rest/during sleep. (sources/channelopathies-jaha-2025)
Quantitative Annual Event Rates
- QTc-stratified risk (ages 18–40, n=812): QTc <460 ms: <0.1%/yr SAE (very low); QTc 470–499 ms: 0.2–0.4%/yr SAE, CE 31%/22 yr; QTc ≥500 ms: 0.5–1.3%/yr SAE, 1.9–2.1%/yr CE — 2–3× higher risk vs QTc <500 ms even on β-blockers; QTc ≥550 ms: SAE 19%/22 yr. (sources/lqts-jaccep-2022, rating: high)
- By genotype (annual rates, up to age 40): LQT1 SAE 0.2–0.5%, CE 0.8–1.1%; LQT2 SAE 0.3–0.7%, CE 0.8–1.8%; LQT3 SAE 0.3–1.1%, CE 0.5–1.5%. (sources/lqts-jaccep-2022)
- Age-specific peaks and syndromal risk: Peak mortality LQT1 ages 1–19; LQT2 ages 30–39; LQT3 ages 15–19. JLN syndrome: 35% SCD risk by age 40 despite β-blocker therapy. (sources/lqts-jaccep-2022)
Variant-Level Risk
- LQT1 variant-specific: KCNQ1-A341V → 80% CE risk by age 40 (vs 30% for non-A341V LQT1); KCNQ1-Y111C founder variant → very low risk (0.05%/year SAE over 25 years, n=80). (sources/lqts-jaccep-2022)
- LQT3 variant-specific: ΔKPQ → higher risk; E1784K and D1790G → significantly less CE. (sources/lqts-jaccep-2022)
- Variant location hierarchy (consistent evidence): Transmembrane pore variants > other transmembrane domain variants > N/C terminus variants (greatest → lowest relative risk). Dominant-negative effect variants carry greater risk than haploinsufficiency variants. (sources/lqts-jaccep-2022)
Management
- Lifestyle modification: Avoid QT-prolonging drugs (CredibleMeds list); avoid competitive sport in high-risk subtypes; correct hypokalaemia/hypomagnesaemia. (sources/channelopathies-jaha-2025, rating: high)
- Beta-blockers (non-selective): Class I — only two confirmed effective agents: propranolol (2.0–3.5 mg/kg/day) and nadolol (1.0–1.5 mg/kg/day). Metoprolol should NOT be used — higher recurrence of events (Chockalingam 2012). (sources/lqts-nejm-2025, rating: very high; sources/VA-SCD-ESC-2022)
- Beta-blocker dosing and monitoring: Nadolol target dose 1 mg/kg/day (evening or divided doses). Monitoring endpoint: 15–20% heart rate blunting during maximal exercise (not QTc reduction). >50% of LQTS patients prescribed β-blockers have suboptimal adherence — most beta-blocker "failures" are due to non-adherence or co-administration of QT-prolonging drugs. (sources/lqts-jaccep-2022, rating: high; sources/lqts-nejm-2025)
- Mexiletine (Na⁺ channel blocker) in LQT3 with prolonged QTc: Class I (upgraded from IIa in ESC 2022). Mexiletine 8 mg/kg/day → 60 ms QTc reduction in LQT3 while reducing CE. In LQT2: ~70% of patients show clinically meaningful QTc shortening with mexiletine — substantially broadening its use beyond LQT3. Acute oral drug test: 6–8 mg/kg dose → measure QTc at 2 hours; shortening >40 ms = positive response; only start long-term therapy in responders. (sources/lqts-nejm-2025, rating: very high; sources/VA-SCD-ESC-2022, sources/lqts-jaccep-2022, sources/scn5a-jaccep-2018)
- ICD + beta-blockers after cardiac arrest: Class I. ICD if symptomatic on beta-blockers + genotype-specific therapies: Class I. (sources/VA-SCD-ESC-2022)
- ICD complication rates (meta-analysis, n=462 LQTS patients with ICD): 2.8%/year inappropriate shocks; 7.0%/year total complications (lead malfunction, device infection, psychological). Dual-chamber transvenous ICD preferred over S-ICD in LQTS — enables atrial overdrive pacing. S-ICD experience limited (EFFORTLESS registry). (sources/lqts-jaccep-2022)
- Primary prevention ICD thresholds: Independent predictors of appropriate shock: QTc ≥500 ms; cardiogenic syncope despite β-blockers. JLN/compound heterozygotes <40 years: shared decision-making re primary prevention ICD or LCSD. (sources/lqts-jaccep-2022)
- LCSD (left cardiac sympathetic denervation): Class I — when ICD is contraindicated/declined, OR patient on full therapy with ICD still has multiple shocks or syncope from VA. Post-LCSD QTc <500 ms predicts success; QTc >500 ms persistent = consider ICD. In certain high-risk LQT1 patients, may be preferred over primary prevention ICD. See concepts/Left-Cardiac-Sympathetic-Denervation. (sources/VA-SCD-ESC-2022, sources/lqts-jaccep-2022)
Acquired Long QT Syndrome
- Caused by IKr-blocking drugs (>200 drugs), hypokalemia, bradycardia, and heart block; individual repolarisation reserve (genetically modulated) determines susceptibility
- Three predictors of latent congenital LQTS in patients presenting with acquired LQTS: (1) age <40 years; (2) QTc >440 ms at baseline (before offending drug); (3) arrhythmic episodes — if any present, offer molecular genetic testing (sources/lqts-nejm-2025, rating: very high)
- Rare variants consistently associated with acquired LQTS: KCNE1-p.D85N and SCN5A-p.S1103Y — testing for these can be considered in patients with acquired LQTS (sources/lqts-nejm-2025)
- 61 common genetic variants collectively explain up to 30% of variability in acquired LQTS (research setting only; routine clinical polygenic testing not yet recommended) (sources/lqts-nejm-2025)
- Excessive physical training can induce marked QT prolongation mimicking congenital LQTS, especially in teenagers; these patients are asymptomatic, genotype-negative, and have no family history; abnormalities are reversible with 3–4 months detraining (sources/lqts-nejm-2025)
Precision Therapy in Practice (Mayo Clinic 20-Year Cohort, n=1,304)
Real-world expert management used up to 18 distinct configurations for LQT3 alone; phenotypic expression outweighs genotype in guiding treatment. (sources/precision-lqts-tcm-2024, rating: high)
- Intentional nontherapy (18%): Post-pubertal, asymptomatic patients with normal resting QTc and normal recovery QTc on stress testing. Precautionary measures (QT-drug avoidance, electrolyte correction, fever control) replace active therapy. Zero lethal events at mean 7.5 years follow-up. (sources/precision-lqts-tcm-2024)
- Genotype-specific prescribing: Propranolol preferred over nadolol in LQT3 — propranolol directly inhibits INaLate, nadolol does not. Mexiletine (mean QTc reduction 65 ms, zero BCEs at 6.5 yr) and IPAP (target ≥80 bpm; BCE rate 1.01 → 0.02/yr) for high-risk LQT2. Flecainide for p.Ile1768Val-SCN5A (variant accelerates Nav1.5 recovery from inactivation — matched to mechanism). (sources/precision-lqts-tcm-2024)
- Escalation ladder: LCSD monotherapy (5%; primarily BB-intolerant; zero lethal events). RCSD added incrementally in refractory LQT3 on top of LCSD + ICD. Cardiac transplant in 4 LQT3 patients (2.6%) with intractable arrhythmia and multiple ICD shocks; continuous IV lidocaine as bridge. (sources/precision-lqts-tcm-2024)
- ICD selection and over-implantation: SQ-ICD avoided in LQT2 (T-wave oversensing); epicardial ICD for high-velocity collision sports; never implanted solely to enable sports participation. 9.1% of referred patients carried an ICD at first evaluation; >50% underwent extraction after specialist reassessment — systematic over-implantation at non-specialist centres. (sources/precision-lqts-tcm-2024)
- See concepts/Precision-Medicine-LQTS for full genotype-specific configurations and outcomes.
Special Populations
Pregnancy and Postpartum
- Risk paradox: Pregnancy is relatively protective (physiological tachycardia shortens QT), but the 9-month postpartum period carries a 2.7-fold increased cardiac event risk and 4.1-fold life-threatening event risk versus prepregnancy baseline (Seth 2007). Risk reverts to baseline after 9 months postpartum. (sources/lqts-pregnancy-medicina-2022, rating: medium)
- LQT2 highest postpartum risk: Postpartum cardiac events are disproportionately reported in LQT2, attributable to the postpartum fall in progesterone, which has direct antiarrhythmic properties via reduction of polymorphic VT in preclinical models. (sources/lqts-pregnancy-medicina-2022)
- Beta-blockers: Class I throughout pregnancy and ≥40 weeks postpartum (ESC 2018; AHA/ACC/HRS 2017). Non-selective agents (propranolol, nadolol) superior to metoprolol. Postpartum event rate reduced from 3.7% → 0.8% with beta-blocker use. (sources/lqts-pregnancy-medicina-2022)
- Amiodarone: contraindicated in pregnancy (QT prolongation, fetal toxicity). Mexiletine and ranolazine can be used as add-on in LQT3. (sources/lqts-pregnancy-medicina-2022)
- Delivery risk stratification (ESC 2018 / Roston 2020): Low risk (QTc ≤470 ms, no events) → Level 1 standard obstetric; medium risk (QTc ≥470 ms or remote events) → Level 2, tertiary centre; high risk (recent events on therapy) → Level 3, Caesarean in cardiac theatre. (sources/lqts-pregnancy-medicina-2022)
- ICD before pregnancy preferred in high-risk women; safe to implant during pregnancy if new indication arises (after 8 weeks gestation). (sources/lqts-pregnancy-medicina-2022)
- See concepts/LQTS-Pregnancy-Management for full detail.
Competitive Sports (AHA/ACC 2025)
- Concealed variant-positive LQTS (gene+, QTc <460 ms at rest): Competitive sports participation reasonable. In the largest LQTS sports cohort (n=494 athletes), zero deaths and 0.3 events/100 patient-years were observed. Avoid QT-prolonging drug exposures; consider prophylactic beta-blocker. (sources/competitive-sports-aha-2025, rating: very high)
- Phenotypic LQTS (QTc ≥460 ms prepuberty / ≥470 ms male / ≥480 ms female): Competitive sports reasonable with shared decision-making (SDM) under expert supervision after risk assessment and guideline-directed therapy. Zero deaths reported; 1.16 events/100 athlete-years in largest cohort. Requires non-selective beta-blockers, emergency action plan (EAP) with AED access. (sources/competitive-sports-aha-2025)
- LQT1 swimming/diving: Can consider with SDM and appropriate precautions — supervision by CPR-trained individual, preference for pool over open water, AED on site. Most prior SCA cases were in undiagnosed/untreated patients. (sources/competitive-sports-aha-2025)
- Clinical stability requirement: No breakthrough arrhythmias for ≥3 months before resuming competitive sports. (sources/competitive-sports-aha-2025)
- ICD for the sole purpose of competitive sports participation: should NOT be performed — ~5%/year inappropriate shock risk + ~4%/year ICD-related complications. (sources/competitive-sports-aha-2025)
- See concepts/Sports-Cardiology-SDM for full SDM framework.
Emerging Therapies
- SupRep gene therapy (LQT1): AAV9-delivered shRNA suppresses mutant KCNQ1 + shRNA-immune replacement cDNA. Normalised QTi/APD90 and restored β-adrenergic response in transgenic rabbits (Bains 2024; Dotzler 2023). (sources/gene-therapy-arrhythmia-2025, rating: high)
- SupRep gene therapy (LQT2): Normalised QTc from 470 → 414 ms; suppressed EADs and TdP inducibility in rabbit model (Bains 2023). (sources/gene-therapy-arrhythmia-2025)
- SupRep gene therapy (Calmodulinopathy): SupRep validated in CALM1/2/3-mediated arrhythmia disorders using a single-construct approach (Hamrick 2024) — the mutation-agnostic strategy overcomes the problem of variant-specific silencing across the three calmodulin genes. (sources/lqts-nejm-2025, rating: very high)
- LQT3 base editing: ABE8e-SpRY via dual AAV9 corrected SCN5A-M1875T in mice with 54% editing efficiency; normalised QTc and APD90; reduced late INa by 66%; ~20% editing sufficient to prevent arrhythmias via electrotonic coupling (Qi 2024). (sources/gene-therapy-arrhythmia-2025)
- Lumacaftor (CFTR potentiator): Phase II trial (NCT04581408) to rescue LQT2 phenotype by improving KCNH2 channel trafficking. (sources/channelopathies-jaha-2025)
- Allele-specific RNAi is limited by mutational heterogeneity — SupRep's mutation-agnostic approach overcomes this. High-throughput patch-clamp for VUS evaluation; iPSC-CM + CRISPR for variant-specific drug testing. (sources/gene-therapy-arrhythmia-2025)
Contradictions / Open Questions
- ClinGen revalidation removed many published LQTS genes: The 2020 Adler et al. ClinGen reappraisal confirmed only 3 genes as definitively causing isolated LQTS (KCNQ1, KCNH2, SCN5A via the new broad rhythm disorder designation); KCNE2 was disputed and removed; KCNJ2/KCNE1 reclassified as primarily syndromal; SCN4B disputed. Many previously published "LQTS genes" on broad panels lack sufficient curated evidence — clinicians risk misclassification by reporting variants in these genes as P/LP for LQTS. (sources/arrhythmia-genetics-mgenetik-2025, sources/clingen-summary-2026-05-09, rating: high)
- ClinGen 2026 clinical implications — SCN4B, CAV3, CALM1/2/3: SCN4B (LQT10) is disputed — variants should not be labelled P/LP for LQTS. CAV3 and KCNJ2 have only limited LQTS evidence — relevant for Andersen-Tawil syndrome, not isolated LQTS interpretation. CALM1/2/3 are definitively established — neonatal LQTS or extremely prolonged QTc without other cause should always include calmodulin gene sequencing. TRDN has strong evidence for a recessive LQTS/CPVT5 phenotype — important for recessive unexplained neonatal arrhythmia. (sources/clingen-summary-2026-05-09, rating: high)
- Drug-induced TdP and latent LQTS — discrepant estimates: The AHA 2020 statement reports ~30% of patients with drug-induced QT prolongation carry pathogenic variants in 1 of the 5 major LQTS genes — supporting drug-induced TdP as frequently unmasking subclinical LQTS. In contrast, the arrhythmia genetics review (2025) cites only 10–15%. The discrepancy likely reflects different patient populations, gene panels, and QT prolongation vs. TdP definitions. Neither figure currently supports routine pre-prescription genetic screening. (sources/drug-arrhythmia-aha-2020, sources/arrhythmia-genetics-mgenetik-2025)
- QTc threshold inconsistency — diagnosis vs. clinical practice: ESC 2022 uses QTc ≥480 ms on repeated ECGs as a Class I diagnostic criterion. Many programmes and older guidelines use QTc >450 ms (males) or >470 ms (females). The Schwartz Score uses ≥480 ms for 3 points but overall diagnosis can be made at lower values. This creates real-world variation in who receives a diagnosis and genetic testing referral. (sources/VA-SCD-ESC-2022, sources/channelopathies-jaha-2025)
- Asymptomatic LQTS — ICD threshold undefined: For asymptomatic patients on full beta-blocker therapy with persistently prolonged QTc, ICD implantation is only Class IIb (1-2-3 LQTS Risk calculator guided). No Class I or IIa threshold exists, leaving clinicians without clear guidance for the largest group of LQTS patients. (sources/VA-SCD-ESC-2022)
- LQT1 and LQT2 genotype frequency discrepancy across sources: Krahn 2022 reports KCNQ1 (LQT1) at 40–45% and KCNH2 (LQT2) at ~40% of genotype-positive cases. The channelopathies-jaha-2025 review reports LQT1 at 30–35% and LQT2 at 25–30%. These differences likely reflect selection bias in reported cohorts, with older registry data (International LQTS Registry) enriched for symptomatic and severe phenotypes, while more recent genetic screening cohorts capture a broader spectrum. The practical implication is that LQT1 and LQT2 together account for 70–85% of genotype-positive LQTS regardless of source. (sources/lqts-jaccep-2022, sources/channelopathies-jaha-2025)
- LQT3: gene therapy vs. Class I mexiletine — competing strategies: ESC 2022 upgraded mexiletine to Class I for LQT3 with prolonged QTc (established pharmacology; long-term registry n=34). Simultaneously, SCN5A base editing shows strong preclinical data. Both address the same INaL mechanism but compete as clinical strategies; no comparative efficacy data exist. (sources/VA-SCD-ESC-2022, sources/gene-therapy-arrhythmia-2025, sources/scn5a-jaccep-2018)
- SupRep balance constraint — no validated human dosing: SupRep requires a precise suppression-to-replacement ratio: excess suppression worsens LQTS; excess replacement risks SQTS. This therapeutic window has been demonstrated in animal models but has never been established in humans. (sources/gene-therapy-arrhythmia-2025)
- NOS1AP biology paradox — partially resolved: NOS1AP risk alleles (associated with longer QT and higher arrhythmic risk in humans) correlate with higher NOS1AP expression in human ventricular myocardium. However, NOS1AP overexpression in guinea pig and rat ventricular myocytes shortens APD — opposite to humans. Dababneh 2025 confirmed NOS1AP modulates repolarisation via NOS1 activity in hiPSC-CMs, establishing a mechanistic pathway, but the directional discrepancy between standard animal models and human data remains unreconciled. (sources/modifier-genes-scd-ehj-2018, sources/gwas-arrhythmias-cmp-genes-2025)
- LQT3 beta-blocker initial uncertainty — reversed by evidence: Early clinical and preclinical data suggested beta-blockers were ineffective or potentially harmful in LQT3 (mechanistic reasoning: LQT3 events at rest → beta-blocker-induced bradycardia could worsen QT prolongation). Later studies demonstrated clear benefit. Caution against extrapolating mechanism-based predictions to clinical outcomes without long-term data. (sources/scn5a-jaccep-2018)
- ICD overimplantation vs. underimplantation tension: The Mayo Clinic cohort shows >50% of patients referred with a pre-existing ICD had it extracted after specialist re-evaluation — suggesting systematic overimplantation at non-specialist centres. Yet ICD provides a mortality benefit in LQTS (Wang 2021 JACC). The resolution is that ICD should be individualised and precision-risk-stratified, not reflexively prescribed for any LQTS diagnosis. ICD over-prescribing creates real harms: inappropriate shocks, anxiety, PTSD, device complications. (sources/precision-lqts-tcm-2024, rating: high)
- Risk score application timing — ESC 2022 recommendation may cause excessive ICD use: ESC 2022 recommends applying the 1-2-3 LQTS Risk calculator at the initial clinical visit (before therapy initiation) to guide ICD decisions. A 946-patient study (Dusi 2024) showed that if ICD decisions had been made based solely on that risk score at initial visit, 142 ICDs would have been indicated; instead, only 22 were implanted (with yearly therapeutic reassessment). Only 3 patients with an ICD received an appropriate shock; no patient died. A separate 2861-patient study confirmed that only a minority of guideline ICD candidates actually needed a device. Because initiating therapy modifies arrhythmic risk, a decision to implant an ICD before reassessment after therapeutic optimisation is not justifiable. (sources/lqts-nejm-2025, rating: very high; sources/VA-SCD-ESC-2022)
- Mexiletine in LQT2 — off-label but evidence-supported: Guideline-directed therapy restricts mexiletine to LQT3. The Mayo Clinic experience shows mexiletine reduced QTc by 65 ms and eliminated BCEs in a high-risk LQT2 subset with baseline QTc ~543 ms, suggesting late sodium current plays a clinically actionable role in severe LQT2. This challenges the strict genotype-to-drug assignment and supports functional-mechanism-guided prescribing. (sources/precision-lqts-tcm-2024, sources/scn5a-jaccep-2018)
Connections
- Related to entities/KCNQ1
- Related to entities/KCNH2
- Related to entities/SCN5A
- Related to entities/NOS1AP
- Related to entities/Andersen-Tawil-Syndrome
- Related to concepts/Cardiac-Action-Potential
- Related to concepts/Cardiac-Repolarization
- Related to concepts/Torsades-de-Pointes
- Related to concepts/Schwartz-Score
- Related to concepts/Left-Cardiac-Sympathetic-Denervation
- Related to concepts/Modifier-Genes
- Related to concepts/Polygenic-Risk-Score
- Related to concepts/GWAS-Cardiac-Genetics
- Related to concepts/iPSC-Derived-Cardiomyocytes
- Related to concepts/SupRep-Therapy
- Related to concepts/AAV-Gene-Delivery
- Related to concepts/CRISPR-Cas9-in-Channelopathies
- Related to concepts/ClinGen-Gene-Disease-Validity
- Related to concepts/Drug-Induced-Arrhythmia
- Related to concepts/Electrical-Remodeling
- Related to concepts/Sudden-Cardiac-Death
- Related to concepts/Circadian-Rhythm-Cardiac-Electrophysiology
- Related to concepts/LQTS-Pregnancy-Management
- Related to concepts/Sports-Cardiology-SDM
- Related to concepts/Cardiogenetic-Centers
- Related to concepts/Variant-Reclassification
- Related to concepts/Electrical-Storm
- Related to concepts/Epigenetics-Cardiac-Arrhythmia
- Related to concepts/Calmodulinopathy
Sources
- sources/lqts-nejm-2025
- sources/channelopathies-jaha-2025
- sources/arrhythmia-genetics-mgenetik-2025
- sources/drug-arrhythmia-aha-2020
- sources/VA-SCD-ESC-2022
- sources/precision-lqts-tcm-2024
- sources/genetics-va-fcvm-2022
- sources/scn5a-jaccep-2018
- sources/membrane-potential-physrev-2021
- sources/repolarisation-jaccep-2023
- sources/circadian-scd-jmcc-2025
- sources/lqts-pregnancy-medicina-2022
- sources/modifier-genes-scd-ehj-2018
- sources/gwas-arrhythmias-cmp-genes-2025
- sources/gene-therapy-arrhythmia-2025
- sources/competitive-sports-aha-2025
- sources/genetic-test-aha-2020
- sources/clingen-summary-2026-05-09
- sources/CALM-FCVM-2018
- sources/lqts-jaccep-2022