Congenital Long QT Syndrome
Authors, Journal, Affiliations, Type, DOI
- Authors: Andrew D. Krahn, Zachary Laksman, Raymond W. Sy, Pieter G. Postema, Michael J. Ackerman, Arthur A.M. Wilde, Hui-Chen Han
- Journal: JACC: Clinical Electrophysiology, Vol. 8, No. 5, 2022, pp. 687–706
- Affiliations: University of British Columbia (Krahn, Laksman, Han); University of Sydney (Sy); Amsterdam UMC (Postema, Wilde); Mayo Clinic (Ackerman); Monash University (Han)
- Type: State-of-the-art review
- DOI: https://doi.org/10.1016/j.jacep.2022.02.017
Overview
This state-of-the-art review by leading LQTS experts provides a practical framework for the diagnosis, risk stratification, and management of congenital LQTS, with focus on the three major autosomal dominant subtypes — LQT1 (KCNQ1), LQT2 (KCNH2), and LQT3 (SCN5A). The review is distinguished by its quantitative provocation testing thresholds, detailed QTc-stratified annual event rates, and a nuanced discussion of genotype-specific management. Key clinical messages include that ~25% of apparent "acquired" LQTS harbour a pathogenic variant, JLN syndrome carries a 35% SCD risk by age 40 despite β-blocker therapy, and >50% of patients on β-blockers have suboptimal adherence. The management section emphasises shared decision-making, beta-blocker monitoring via heart rate blunting rather than QTc change, and a step-up approach from conservative measures through pharmacological therapy, LCSD, and ICD.
Keywords
Long QT syndrome, LQTS, LQT1, LQT2, LQT3, KCNQ1, KCNH2, SCN5A, torsades de pointes, risk stratification, beta-blockers, mexiletine, LCSD, ICD, Schwartz score, QTc interval, provocation testing, Jervell and Lange-Nielsen
Key Takeaways
Pathophysiology
- K⁺ channel LOF: KCNQ1 (IKs, LQT1) and KCNH2 (IKr, LQT2) loss-of-function variants reduce phase 3 outward K⁺ current → delayed repolarization → prolonged QT. KV7.1 and KV11.1 account for ~80% of all genotype-positive LQTS cases.
- Na⁺ channel GOF: SCN5A (LQT3) gain-of-function → persistent late INa during plateau → delayed repolarization → QT prolongation.
- β-adrenergic effects by subtype: In LQT1, sympathetic stimulation fails to augment IKs normally → uncompensated APD lengthening. In LQT2, β-stimulation increases depolarising currents but cannot increase repolarising IKr → net APD lengthening. In LQT3, β-stimulation increases late INa. All three converge on EAD generation → TdP trigger.
- Final pathway: EADs → triggered TdP → ventricular fibrillation.
Epidemiology
- Prevalence ~1:2,000 based on newborn evaluation cohort; slight female predominance.
- Up to 40% of genotype-positive LQTS patients have a baseline QTc within the normal range.
- 1–10% of the general population may have QTc ≥450 ms.
- LQTS accounts for 5–10% of young SCD cases at autopsy and ~15% of "unexplained" resuscitated cardiac arrest.
Diagnosis
Guideline Criteria
- LQTS is definitely diagnosed with: Schwartz score ≥3.5, a pathogenic variant, or repeated QTc ≥500 ms in absence of QT-prolonging drugs (HRS/EHRA/APHRS consensus).
- Schwartz score ≥3.5: 99% specificity, 19–36% sensitivity.
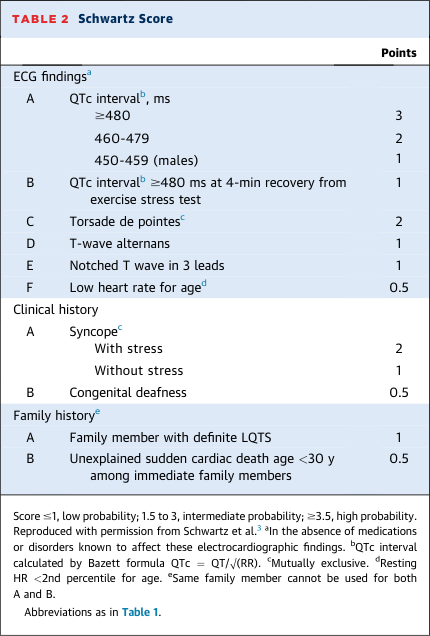
- Updated Schwartz score incorporates QTc ≥480 ms at 4-minute exercise recovery as a scoring criterion.
- Important caveat: ~25% of patients with apparent "acquired" LQTS harboured a pathogenic variant in an international cohort — all patients with drug-induced QT prolongation should be evaluated after drug cessation.
ECG
- QTc thresholds: ≥450 ms in men, ≥460 ms in women (absence of QT-prolonging agents) → increased pretest probability.
- Best measurement lead: Lead II or V5; V2/V3 provide longer QTc measurements but lower predictive power for variant carriers.
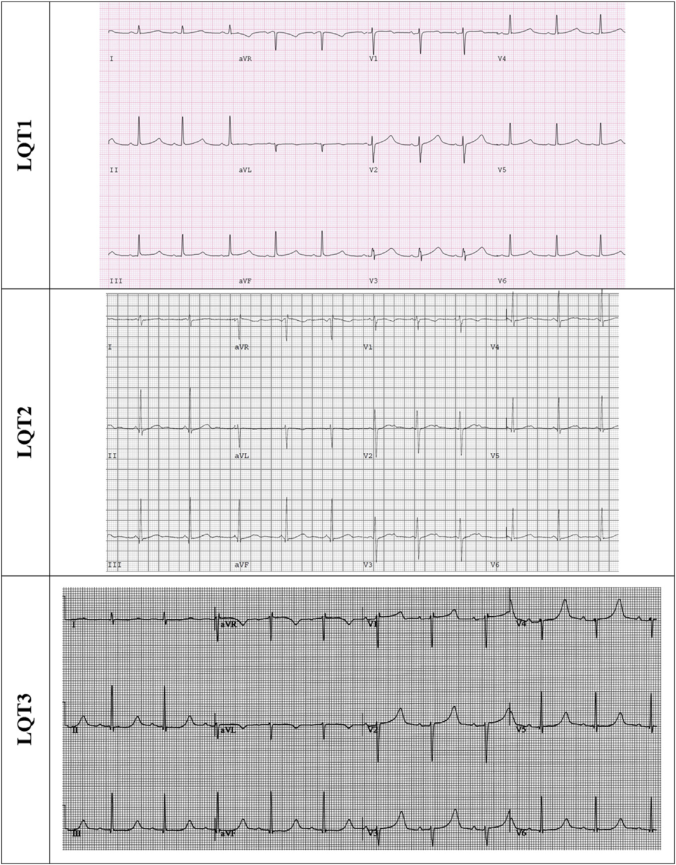
- T-wave patterns by genotype: LQT1 → broad-based T-wave; LQT2 → low-amplitude bifid T-wave; LQT3 → late-onset peaked or asymmetric biphasic T-wave. Giant T-waves (T-U waves) herald TdP.
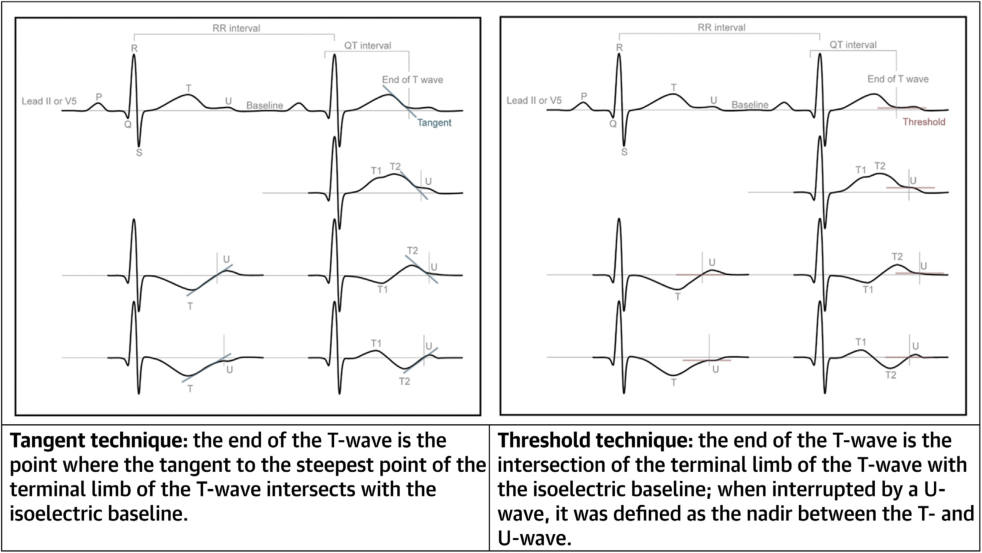
- Bazett formula (QTc = QT/√RR) remains the standard despite overestimating QTc.
Provocation Testing
- Exercise testing (QTc at 4-min recovery):
- ≥445 ms → 90% sensitivity and 90% specificity for LQTS diagnosis (incorporated into updated Schwartz score)
- ≥480 ms → 36% sensitivity and 100% specificity
- LQT1: QTc increases during exercise, gradually shortens during recovery; LQT2: QTc shortens during exercise, lengthens during recovery — useful for genotype differentiation.
- Validated for LQT1 and LQT2 only; children require 7-min recovery (QTc >460 ms optimal cutoff).
- Stand-up (orthostatic) test:
- QTc stretch ≥490 ms (maximal QT stretch = T-wave end approaches next P-wave) → 92% sensitivity, 79% specificity
- Not validated in paediatric patients (healthy children can increase QTc up to 80 ms on standing).
- Epinephrine challenge: Variable heart rate response limits utility; stand-alone diagnostic value limited; reserved for patients who cannot exercise.
- Mental stress testing requires standardisation; not yet clinically validated.
Genetic Testing
- Recommended for Schwartz score ≥3; yield 50–80% depending on pretest probability.
- Major gene frequencies (genotype-positive LQTS): KCNQ1 40–45% (LQT1), KCNH2 ~40% (LQT2), SCN5A 5–10% (LQT3).
- "Minor" LQTS genes (CAV3, KCNE1, KCNE2, KCNJ2, KCNJ5, SCN4B) now considered "limited evidence" per ACMG reappraisal.
- Polygenic influences and periodic variant reinterpretation are relevant; genetics is ever-evolving.
Family Screening
- Cascade screening of all first-degree relatives is mandatory.
- If proband has an identified variant → targeted genetic testing in first-degree relatives; carriers undergo clinical assessment (ECG + exercise testing).
- If proband is gene-elusive → all first-degree relatives undergo clinical assessment.
- Baseline echocardiogram recommended in all patients assessed for LQTS to exclude structural heart disease.
Risk Stratification
Clinical Factors
- Age: Greatest risk in childhood; attenuates over time; >60 years: competing mortality from other conditions.
- Sex-age interaction:
- Males aged 10–12: 4.0× relative risk of SAE vs females (Hobbs cohort, n=2,272).
- Males: 2.3× relative risk of SAE until age 13 (Costa cohort, n=1,051 LQT1).
- Adults age 18–40: CE rate 40% female vs 14% male; SAE rate 11% female vs 2% male (Sauer, n=812).
- Mechanism: testosterone potentiates IKs; oestrogen inhibits IKr; progesterone potentiates IKs.
- Syncope: ≥2.0× relative risk of SAE; greater risk if syncope occurred within 2 years or is recurrent.
QTc Interval
- Annual rates by QTc stratum (ages 18–40, Sauer n=812):
- QTc ≤439 ms: CE 3%, SAE 0%
- QTc 440–469 ms: CE 23%, SAE 2%
- QTc 470–499 ms: CE 31%, SAE 4%
- QTc 500–549 ms: CE 41%, SAE 12%
- QTc ≥550 ms: CE 46%, SAE 19%
- QTc ≥500 ms: 1.9–2.1%/year CE; 0.5–1.3%/year SAE; 2–3× higher risk than QTc <500 ms even on β-blockers.
- QTc <500 ms: 0.2–0.4%/year SAE.
- QTc <460 ms: <0.1%/year SAE — very low risk.
- ~1.1× HR for CE/SAE per 10 ms QTc increment.
- Maximum QTc on serial ECGs provides greater CE prediction than single measurement.
Genetic Factors
- Annual SAE rates by genotype (up to age 40):
- LQT1: 0.2–0.5%/year
- LQT2: 0.3–0.7%/year
- LQT3: 0.3–1.1%/year
- Annual CE rates by genotype (up to age 40):
- LQT1: 0.8–1.1%/year
- LQT2: 0.8–1.8%/year
- LQT3: 0.5–1.5%/year
- Age-specific peak mortality: LQT1 = ages 1–19; LQT2 = ages 30–39; LQT3 = ages 15–19.
- Variant-specific risk (LQT1): A341V → 80% CE risk by age 40 (vs 30% for other LQT1 variants); Y111C founder mutation → very low risk (0.05%/year SAE).
- Variant-specific risk (LQT3): ΔKPQ → higher risk; E1784K and D1790G → significantly less CE.
- Variant location hierarchy: Transmembrane pore variants > other transmembrane domain variants > N/C terminus variants (greatest → lowest relative risk of events). Dominant-negative effect variants > haploinsufficiency variants.
- JLN syndrome (autosomal recessive KCNQ1/KCNE1): 35% SCD risk by age 40 despite β-blocker therapy.
- Common SNPs (NOS1AP, KCNH2-K897T) influence CE/SAE risk in LQTS.
Other Risk Factors
- Negative electromechanical window (aortic valve closure − QT interval) independently associated with symptoms in 651 LQTS patients (Sugrue cohort); requires prospective validation.
Management
Conservative
- QT-prolonging drug avoidance: CredibleMeds database (also as smartphone app); includes illicit substances (amphetamine, cocaine, oxycodone, quetiapine).
- Sports: Overall sport-related event rate in treated LQTS appears low based on predominantly paediatric/adolescent cohort studies. Sports participation reasonable in adequately assessed, treated, educated patients. Shared decision-making with patient, family, and sports representatives advised.
- Exception: LQT1 patients must not swim alone (exertional syncope + drowning risk compounded).
- Conservative measures alone may suffice in asymptomatic LQTS with normal QTc (genotype-positive, little/no phenotype).
Beta-Blockers
- Foundational therapy for symptomatic LQTS or significant QTc prolongation; RR 0.4–0.5 for CE and SAE vs untreated.
- Nadolol: Greatest overall risk reduction; effective for both LQT1 and LQT2. Target dose: 1 mg/kg/day. Evening or divided dosing to minimise side effects.
- Propranolol: May offer greater QTc reduction; proposed efficacy predominantly in LQT1 (direct INaLate inhibition of relevance in LQT3 too per precision cohort data).
- Metoprolol/atenolol/bisoprolol: Second-line if non-selective agents not tolerated.
- Monitoring endpoint: Aim for 15–20% heart rate blunting during maximal exercise (rather than QTc shortening) to confirm adequate effect and adherence.
- Adherence problem: >50% of LQTS patients on β-blockers have suboptimal adherence (Waddell-Smith pharmacy dispensing data) — actual relative risk reduction of optimal therapy likely substantially greater than reported.
- β-blockers are safe during pregnancy and reduce maternal risk especially in the postpartum period; continuation is mandatory.
- Some older/shorter QTc patients may not require lifelong β-blocker therapy.
Mexiletine
- Class IB sodium channel blocker; adjunctive pharmacological agent.
- LQT3: Mexiletine 8 mg/kg/day → significant 60 ms QTc reduction while reducing CE.
- LQT2: Significant QTc reductions also reported; warrants further investigation.
- Access threatened by recent "orphan drug" (neuromuscular) designation limiting availability.
Other Emerging Pharmacotherapy
- Nicorandil: Potentiates K⁺ channels → QTc shortening (small cohorts).
- Long-term potassium replacement: QTc shortening (small cohorts).
- Ranolazine: Sodium channel blockade → shortens QTc in LQT3 (small cohorts).
- Lumacaftor-ivacaftor: Potentiates KV11.1 channel trafficking → QTc shortening in 2 LQT2 patients in vivo; Phase II trial ongoing.
Device Therapy (ICD)
- Secondary prevention (resuscitated CA): Class I indication.
- Primary prevention ICD — challenging; requires balancing absolute SCD risk vs device-related complications.
- Independent predictors of appropriate ICD shock (two large retrospective cohorts): QTc ≥500 ms; cardiogenic syncope despite β-blocker therapy.
- JLN/compound heterozygotes: Consider shared decision-making for primary prevention ICD or LCSD in patients <40 years of age.
- ICD complication data (meta-analysis, n=462): 2.8%/year inappropriate shocks; 7.0%/year total complications (lead malfunction, device infection, psychological consequences).
- Device choice: Dual-chamber transvenous ICD preferred — enables atrial overdrive pacing for acute VA episodes and prevention of pause-dependent QT prolongation.
- S-ICD: Limited experience in LQTS; per EFFORTLESS registry, LQTS patients do not appear at increased inappropriate shock risk vs other populations — but current experience is minimal.
- Pacemaker without ICD: Useful for pause-dependent/bradycardia-related TdP; atrial pacing + β-blockers effective in high-risk JLN paediatric patients (small cohort).
Left Cardiac Sympathetic Denervation (LCSD)
- Reduces CE even on top of β-blockers; or as monotherapy in β-blocker intolerant patients.
- Reserved for those intolerant of pharmacological therapy or with breakthrough CE on therapy.
- In certain high-risk LQT1 patients, may be preferred over primary prevention ICD.
- Invasive; high patient satisfaction despite complication risk.
Future Directions
- Autonomic nervous system role in repolarization instability requires further delineation.
- AI/ML T-wave morphology assessment and genetic prediction algorithms for improved LQTS detection.
- Algorithm-based risk stratification models incorporating multiple independent predictors.
- Contemporary management is evolving toward identifying patients who do not require therapy (more benign phenotypes via genetic diagnosis).
- Mexiletine efficacy (CE/SAE reduction) in larger LQT3 and LQT2 cohorts awaited.
- Lumacaftor-ivacaftor, KCNQ1 ion channel antibodies, KCNQ1-SupRep gene therapy, and SGK1 inhibition (LQT3) in development.
Limitations of the Document
- State-of-the-art review (not systematic review or meta-analysis); conclusions reflect expert opinion in addition to cited evidence.
- Risk stratification data predominantly retrospective, from International LQTS Registry (enriched for severe phenotype); annual event rates likely overestimate risk in contemporary cohorts with more asymptomatic, genetically-identified patients.
- Most management evidence applies specifically to LQT1 and LQT2; LQT3 data are more limited.
- Exercise provocation thresholds validated specifically for adults with LQT1 and LQT2; separate paediatric and LQT3 data are sparse.
- ICD experience with S-ICD in LQTS very limited at time of publication.
- Author disclosures: Multiple industry relationships (Abbott, Boston Scientific, Medtronic, Invitae, LQT Therapeutics, Daiichi-Sankyo).
Key Concepts Mentioned
- concepts/Cardiac-Repolarization — ion channel basis of QT prolongation and TdP generation
- concepts/Torsades-de-Pointes — EAD-mediated trigger for VF in LQTS
- concepts/Schwartz-Score — primary diagnostic tool; updated with exercise QTc threshold
- concepts/Left-Cardiac-Sympathetic-Denervation — management escalation option
- concepts/Drug-Induced-Arrhythmia — acquired LQTS often unmasking congenital disease
- concepts/Sex-Differences-in-Channelopathies — sex-age risk interaction
- concepts/LQTS-Pregnancy-Management — postpartum risk and beta-blocker continuation
- concepts/Precision-Medicine-LQTS — genotype-specific and phenotype-guided therapy
- concepts/Sports-Cardiology-SDM — sports participation shared decision-making
Key Entities Mentioned
- entities/Long-QT-Syndrome — main subject of this review
- entities/KCNQ1 — LQT1 gene; 40–45% of genotype-positive cases
- entities/KCNH2 — LQT2 gene; ~40% of genotype-positive cases
- entities/SCN5A — LQT3 gene; 5–10% of genotype-positive cases
Wiki Pages Updated
- wiki/sources/lqts-jaccep-2022.md — created
- wiki/sourceindex.md — entry added
- wiki/entities/Long-QT-Syndrome.md — provocation testing thresholds, annual event rates by QTc/genotype, JLN 35%, ~25% acquired LQTS, ICD complication rates, variant location hierarchy, beta-blocker HR blunting endpoint, source count updated
- wiki/concepts/Schwartz-Score.md — sensitivity/specificity data, exercise QTc incorporation
- wiki/entities/KCNQ1.md — LQT1 annual event rates, A341V/Y111C variant risk data
- wiki/entities/KCNH2.md — LQT2 annual event rates, pore-variant risk hierarchy